Stochastic Simulation and Analysis
Stochastic dynamics at the molecular level play a key role in cell biology. Such dynamics can have subtle dynamic effects that often contribute to biological function in interesting and unexpected ways. Yet stochastic dynamics in living cells have been far less studied than their deterministic counterparts, and fewer tools exist for their more demanding simulation and analysis. We help fill this gap by developing novel computational methods and software for the modeling, simulation, and analysis of stochastic systems in biology.
Solving the Chemical Master Equation
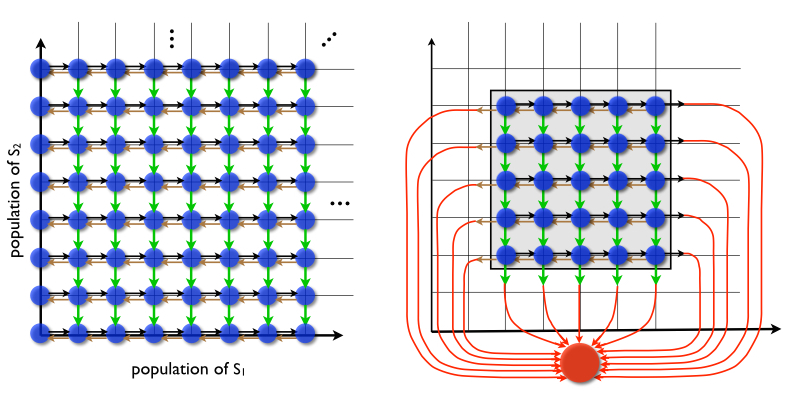
The Chemical Master Equation (CME), describes the time evolution of the joint probability density function of the various species being modeled in a stochastic chemically reacting system. We have been developing tools for the accurate solution of the CME. The Finite State Projection method, which has been developed by the PI and his former studenty Munsky, provides a means for reducing the infinite CME into a finite linear system of equations. This system could be quite large, however, and one faces the curse of dimensionality when looking at larger problems. Recent work in collaboration with the Schwab group aims at overcoming the curse of dimensionality. It is based on the use of Quantized Tensor Train (QTT) formatted numerical linear algebra for the low parametric, numerical representation of tensors.
V. Kazeev, M. Khammash, M. Nip, and C. Schwab
"Direct solution of the chemical master equation using quantized tensor trains". 2014, PLoS computational biology. external pageDOI
B. Munsky and M. Khammash
"The Finite State Projection Approach for the Analyses of Stochastic Noise in Gene Networks," IEEE Transactions on Automatic Control, and IEEE Transactions on Circuits and Systems I, Joint Special Issue on Systems Biology, Vol. 53, pp. 201-214, 2008. external pageDOI
B. Munsky and M. Khammash
"A Multiple Time Interval Finite State Projection Algorithm for the Solution to the Chemical Master Equation," Journal of Computational Physics, Vol. 226, No. 1, pp. 818-835, 2007. external pageDOI
Slaven Peles, Brian Munsky and Mustafa Khammash
"Reduction and Solution of the Chemical Master Equation Using Time-Scale Separation and Finite State Projection," Journal of Chemical Physics, 125 (22), 503646, December 2006. external pageDOI
Brian Munsky and Mustafa Khammash
"The Finite State Projection Algorithm for the Solution of the Chemical Master Equation," Journal of Chemical Physics, Vol. 124, 044104, 2006. external pageDOI
Stochastic Sensitivity Analysis
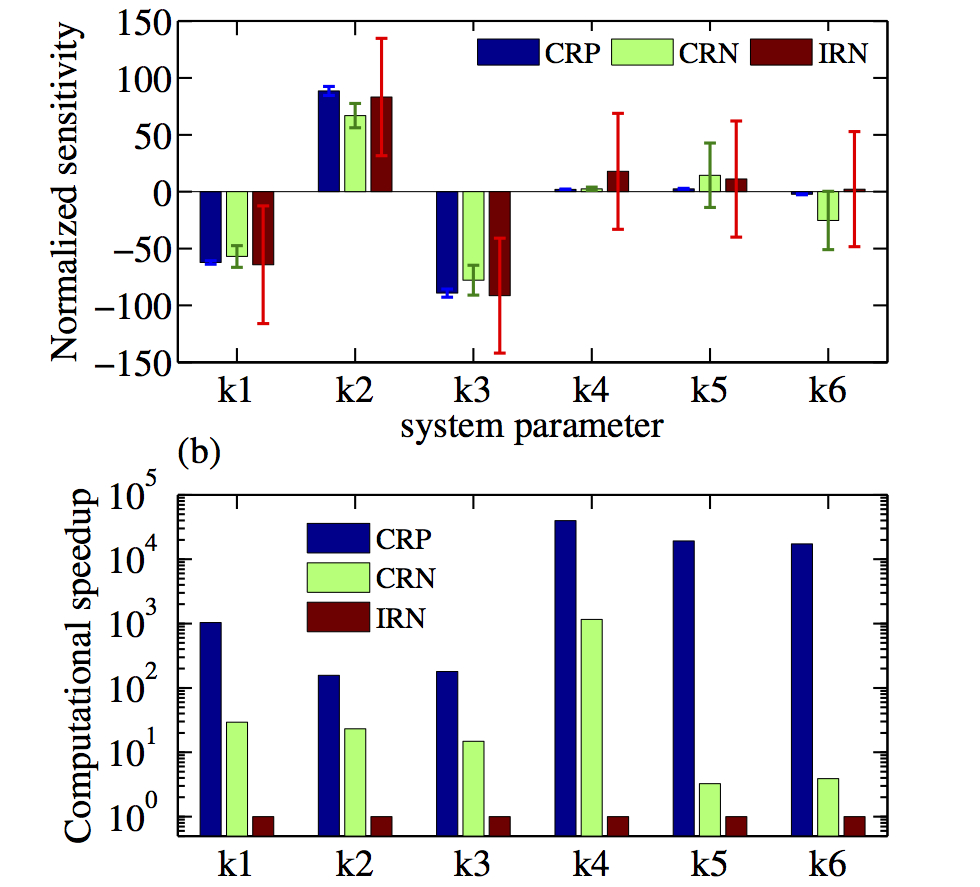
Models of biochemical reaction networks typically involve many kinetic parameters that influence their dynamic behavior. Sensitivity analysis is an indepsensible tool for their study and evaluation. Such analysis quantifies the effect of changes in parameter values on an outcome of interest, giving insight into network design and its robustness properties. Sensitivity analysis is used to identify the important reactions for a given output, which can be used in experiment design and to identify potential targets for drug therapy. Finally sensitivity analysis can aid in the estimation of model parameters and in optimizing and fine-tuning the system’s behaviour in synethic circuit design. We have been developing very efficient computational tools and software for the sensitivity analysis of stochastic models of biochemical reaction networks. By exploiting different couplings of reaction trajectories, we are able to develop methods that are often orders of maganitude more efficient than what what was previously possible.
The existing sensitivity estimation methods become computationally infeasible for even moderately large networks because they rely on time-consuming Monte Carlo simulations of the exact reaction dynamics. Currently we are working towards eliminating this critical bottleneck by developing methods that can accurately estimate parameter sensitivities using only approximate tau-leap simulations of the reaction dynamics. In the future we would like to combine such methods with multilevel strategies to gain computational efficiency and lower the approximation bias in the sensitivity estimator.
A. Gupta and M. Khammash
"An efficient and unbiased method for sensitivity analysis of stochastic reaction networks," J R Soc Interface, external pagein press.
A. Gupta and M. Khammash
"Sensitivity analysis for stochastic chemical reaction networks with multiple time-scales," Electron. J. Probab. 19, no. 59, pp. 1–53, 2014. external pageDOI
A. Milias-Argeitis, J. Lygeros, and M. Khammash
"Fast variance reduction for steady-state simulation and sensitivity analysis of stochastic chemical systems using shadow functions," Journal of Chemical Physics, 141(2):024104, 2014. external pageDOI
A. Gupta and M. Khammash
"Unbiased estimation of parameter sensitivities for stochastic chemical reaction networks", SIAM : Journal on Scientific Computing, 2013. external pageDOI
P. Sheppard, M. Rathinam, and M. Khammash
"SPSens: A software package for stochastic parameter sensitivity analysis of biochemical reaction networks," Bioinformatics, 29(1), 140-142, 2013. external pageDOI
P. Sheppard, M. Rathinam, and M. Khammash
"A pathwise-derivative approach to the computation of parameter sensitivities in discrete stochastic chemical systems", Journal of Chemical Physics, Vol. 136, 034115, 2012. external pageDOI
M. Rathinam, P. Sheppard, and M. Khammash
"Efficient Computation of Parameter Sensitivities of Discrete Stochastic Chemical Reaction Networks," Journal of Chemical Physics, Vol. 132, 034103, January 2010. external pageDOI
Long-range behavior of Stochastic Reaction Networks
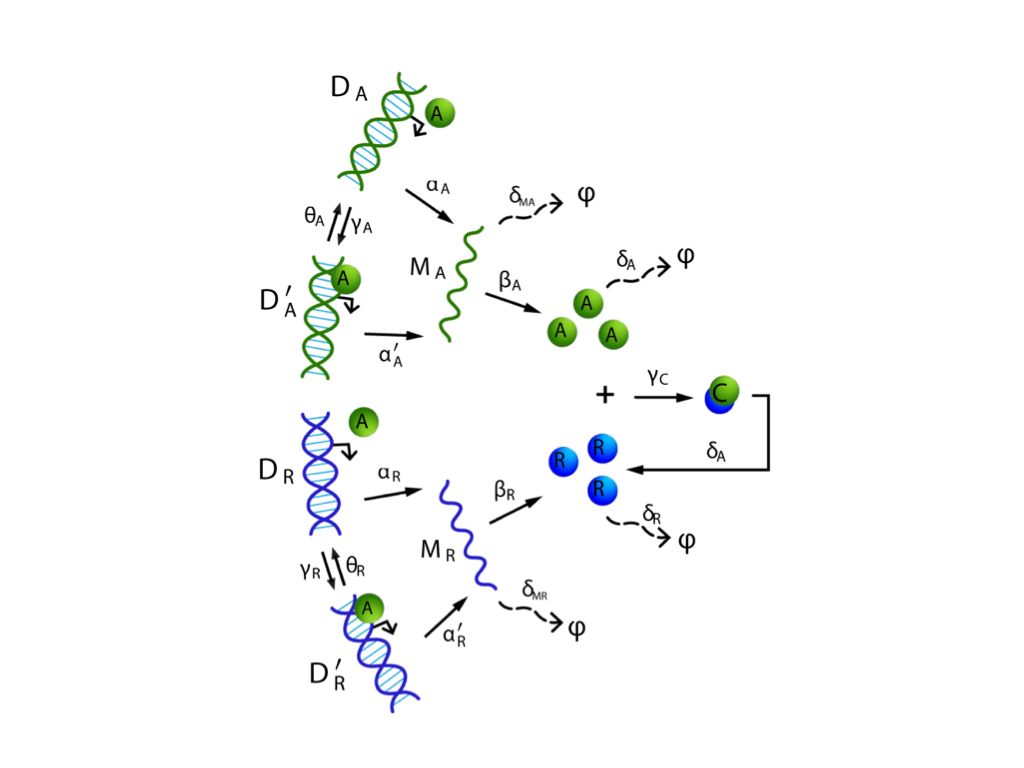
We have been developing a constructive scalable framework for examining the long-term behavior and stability properties of stochastic reaction dynamics. Specifically, we address the problem of determining ergodicity of the reaction dynamics, which is analogous to having a globally attracting fixed point for deterministic dynamics. The ergodicity properties of a process enable us to examine when the statistical moments of that process remain bounded over time and when they converge to their steady state values. The framework we develop relies on a blend of ideas from probability theory, linear algebra and optimization theory. Using this approach, we can show that the stability properties of a wide class of biological networks can be assessed using efficient and scalable linear programs, well-known for their tractability. The validity, efficiency, and wide applicability of these results can be demonstrated on several reaction networks arising in biochemistry, systems biology, epidemiology and ecology.
A. Gupta, C. Briat, and M. Khammash
"A Scalable Computational Framework for Establishing Long-Term Behavior of Stochastic Reaction Networks," PLoS Comput Biol, 10(6): e1003669, 2014. external pageDOI
C. Briat, A. Gupta, I. Shames and M. Khammash
"Scalable tests for ergodicity analysis of large-scale interconnected stochastic reaction networks", 21st International Symposium on Mathematical Theory of Networks and Systems, Groningen, The Netherlands, 2014.
A. Gupta and M. Khammash, “Determining the long-term behavior of cell populations: A new procedure for detecting ergodicity in large stochastic reaction networks”, IFAC World Congress 2014, Volume 19(1).
Hybrid Simulation Methods
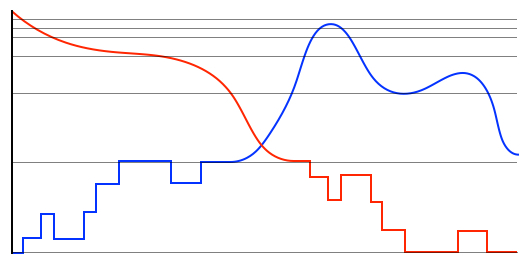
Hybrid methods for simulating a system of stochastically reacting species typically partition the set of species and set of reactions into discrete and continuous subsets, then rely on quasi-stationary assumptions to approximate the dynamics of fast subnetworks. As the dynamics evolve in time these partitions might have to be adapted during the simulation. We have recently been developing hybrid methods that automatically partition the reactions and species sets into discrete and continuous components, while applying dynamically the quasi-stationary assumption. Our methods do not require user intervention, as they exploit the changing timescale separation between reactions and/or changing magnitudes of copy-numbers of constituent species. Application to problems in Systems Biology show that very good approximations can be achieved in significantly less computational time. These tools are especially suitable for systems with oscillatory dynamics where the system dynamics change considerably throughout the time-period of interest.